
Dirbtinio intelekto programos „AlphaFold“ pasiekimai priverčia išsižioti, tačiau turi ir savų apribojimų. „Programa „AlphaFold“ išsprendė problemą, kuri atrodė neišsprendžiama – ji neeksperimentiniais būdais nustato baltymų ir baltymų kompleksų struktūras. Programa tai tikrai daro labai gerai ir tiksliai. Laimei, „AlphaFold“ dar nėra visagalis, nes jeigu galėtų viską nustatyti, mes liktume be darbo“, – juokaudamas pasakoja Vilniaus universiteto Gyvybės mokslų centro biochemikas dr. Giedrius Sasnauskas.
Mokslininkas dr. G. Sasnauskas – vienas iš nedaugelio struktūrinės biologijos tyrėjų Lietuvoje. Kaip pats sako, žvelgdamas į atominius pasaulius, mato tai, ko niekas nematė iki tol. Smalsumas nagrinėti nanometrų dydžio trimačius tankius labirintus primenančius objektus dr. G. Sasnauską atvedė iki jo ir kolegų sėkmės istorijos – nustatytos genų redagavimo įrankio CRISPR-Cas pirmtako struktūros.
– Kuo jums įdomi struktūrinė biologija ir kaip jai gali tarnauti jūsų naudojamas krioelektroninės mikroskopijos metodas?
– Vieni žmonės žiūri į žvaigždes, ieško to, ko dar niekas nebuvo atradęs. Kiti gal keliauja į kalnus, tolimus miškus, ieško niekam nematyto augalo ar vabalo. O mes, struktūrinės biologijos mokslininkai, keliaujame į nanometrais, tai yra milijonosiomis milimetro dalimis, matuojamą pasaulį ir esame pirmieji, kurie pamatome, kaip atrodo gyvuosius organizmus sudarančios molekulės. Tame matau ne mažesnį žavesį ir net azartą.
Plačiausia prasme struktūrinė biologija yra mokslas apie biologines struktūras – nuo plika akimi ar įprastu šviesiniu mikroskopu matomų dalykų, kaip atskiros ląstelės ar ląstelių dalys, iki daug mažesnių objektų – pavyzdžiui, atskirų molekulių.
Šiuolaikinė struktūrinė biologija daugiausia tiria būtent gyvuosius organizmus sudarančių molekulių, ypač baltymų, sandarą atomų lygmeniu. Tai yra nustatome, kaip erdvėje išsidėsto tūkstančiai tiriamo baltymo molekulę sudarančių atomų. Tipinis mūsų tyrimų objektas yra nuo kelių nanometrų iki šimto nanometrų dydžio – vadinasi, nuo atskiro baltymo iki nedidelio viruso.
Struktūrinėje biologijoje naudojami trys pagrindiniai eksperimentiniai metodai – branduolių magnetinis rezonansas, rentgeno spindulių kristalografija ir kriogeninė elektroninė mikroskopija, arba trumpai krio-EM. Būtent krio-EM, nors ir vėliausiai atsiradęs, dėl savo efektyvumo ir patogumo šiuo metu tampa plačiausiai naudojamu baltymų struktūroms nustatyti skirtu metodu. Gavę Europos Sąjungos fondų finansavimą kriogeninį elektroninį mikroskopą turime Vilniaus universiteto Gyvybės mokslų centre, jį intensyviai naudojame jau trejus metus. Juo galime analizuoti stambius baltymų kompleksus ir net virusines daleles.
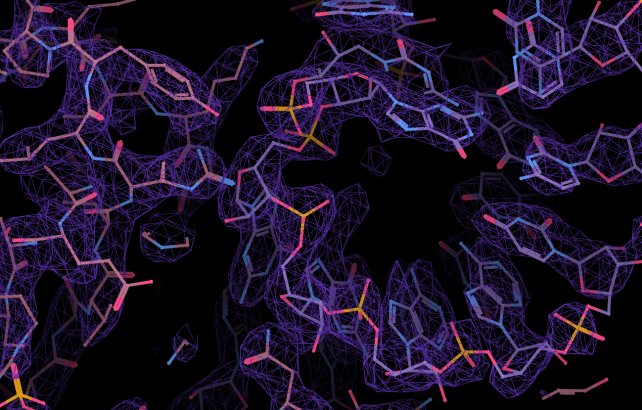
Viruso uodegėlės krio-EM žemėlapis
– Kaip gaunate baltymo struktūrą, naudodami krio-EM mikroskopiją?
– Įdomus kriogeninei mikroskopijai skirtų mėginių paruošimas. Mus dominančios molekulės, pavyzdžiui, baltymo, tirpalas yra užšaldomas kaip labai plonos kelių dešimčių nanometrų storio ledo plokštelės. Tame lede įšąla ir baltymo molekulės. Įsivaizduokite skaidrų kelių centimetrų storio ežero ledą su jame įšalusiais lapais. Kažką panašaus, tik milijoną kartų sumažintą, turime ir mes.
Tada elektroniniu mikroskopu fotografuojame daug šių plonyčių ledo plokštelių, o iš gautų vaizdų išrenkame lede įšalusių atskirų baltymo dalelių vaizdus. Ir iš daugybės, dešimčių ar šimtų tūkstančių, kartais – net milijonų, tokių dalelių atvaizdų gauname labai detalų baltymo molekulės atvaizdą, panašų į tankų trimatį labirintą. Vėliau „klajojame“ po tą labirintą ir jį sudarančius „urvus“ užpildome baltymą sudarančiais atomais. Taip gauname molekulės struktūros modelį.
Esminis krio-EM privalumas, lyginant su tradicine elektronine mikroskopija, yra tas, kad lede įšalęs baltymas ar kita molekulė išlieka savo natūralios „šlapios“ būsenos. Užšaldymas vyksta labai staigiai, todėl pavyksta „pagauti“ baltymą tokį, koks jis yra tirpale. Paprasto elektroninio mikroskopo mėginiai yra sausi, taigi smulkios struktūrinės baltymų detalės jame yra prarandamos. O su krio-EM mes pamatome viską.

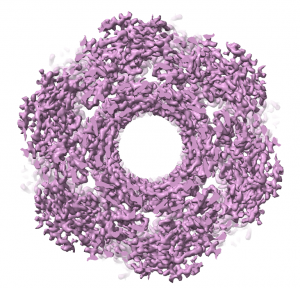
Viruso kapsidės krio-EM žemėlapis
– Dirbtinis intelektas taip pat padeda atskleisti baltymų struktūras. Gana daug dėmesio susilaukia „AlphaFold“ programa. Skelbiama, kad ja baltymų struktūras galima sudaryti itin greitai. Ką jūs manote apie „AlphaFold“? O gal ją naudojate?
– Pastaruosius 15 metų mes, struktūrinės biologijos mokslininkai, tikrai įdomiai gyvenome. Per tą laiką įvyko keli virsmai.
Pirma, vienas iš klasikinių struktūrinės biologijos tyrimų metodų – kristalografija tapo kur kas prieinamesnė daugeliui vartotojų. Antra, įvyko vadinamoji „rezoliucijos revoliucija“ elektroninėje mikroskopijoje, kai vietoje žemos skiriamosios gebos, arba mažai detalių, molekulių atvaizdų, primenančių vieną ar kelias sulipusias bulves, mokslininkai jau galėjo pamatyti smulkesnes detales.
Paskutinis virsmas buvo dirbtiniu intelektu paremtas algoritmas „AlphaFold 2“. Paskelbtas 2021 m. viduryje, jis revoliuciją sukėlė vos ne per vieną dieną. Daugelis žmonių su dirbtinio intelekto galimybėmis susidūrė tik atsiradus „ChatGPT“, o mes, su baltymų struktūromis dirbantys mokslininkai, dirbtinio intelekto jėgą savo kailiu pajutome gerokai anksčiau.
Ką padarė „AlphaFold 2“? Išsprendė problemą, kuri iki tol atrodė neišsprendžiama – jis generuoja labai tikslius atskirų baltymų ar baltymų kompleksų – susijungusių kelių baltymų molekulių – struktūrų modelius, tūkstančius atomų erdvėje sudėlioja angstremo tikslumu.
Taigi „AlphaFold“ atlieka tą patį, ką ir mes, struktūriniai biologai, darome naudodami krio-EM ir kitus eksperimentinius metodus.
Pirmą kartą panaudoję „AlphaFold“ likome išsižioję – modelio ir mūsų pačių nustatytos baltymo struktūros atitikimas buvo toks geras… (juokiasi). Paskui prie to pripratome. Dabar programą naudojame kaip pagalbinę priemonę savo tyrimams.
Laimei, „AlphaFold“ dar nėra visagalis. Kadangi algoritmas buvo apmokytas iš eksperimentinių baltymų struktūrų, būtent daugelio baltymų struktūras jis sėkmingai ir nuspėja. Tačiau dažnai baltymai veikia ne vieni, o susijungę su įvairiais nebaltyminiais komponentais – pavyzdžiui, nukleorūgštimis RNR ir DNR, angliavandeniais ar mažesnėmis molekulėmis. Šių komponentų ir jų sąveikos su baltymais „AlphaFold“, bent kol kas, nemodeliuoja. Čia išlieka reikalingi visi eksperimentiniai metodai, tad darbo tebeturime ir mes. Tačiau neabejoju, kad laikui bėgant „AlphaFold“ ir panašūs algoritmai sugebės vis daugiau.
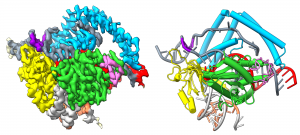
Cas12m baltymo-RNR-DNR komplekso krio-EM žemėlapis ir struktūros modelis
– Norėčiau pakalbėti apie jūsų atradimų pritaikymą. Išsiaiškinome baltymų struktūrą – kur tai galima panaudoti toliau?
– Juokais galima teigti, kad, nustačius baltymo struktūrą, svarbiausia paruošti gražius jos paveikslėlius, kuriais galima „papuošti“ mokslinę publikaciją. Baltymų struktūros išties yra savaip gražios, gali tapti net meno objektais. O kalbant rimčiau, jei sužinome, kaip sudarytas konkretus baltymas, galime suprasti, kaip jis veikia. Kaip katalizuoja kokią nors cheminę reakciją ar atlieka kitą funkciją.
Savo hipotezėms apie baltymo veikimą patikrinti galime suplanuoti ir atlikti įvairius eksperimentus laboratorijoje. O jei suprantame, kaip baltymas veikia, galime pagalvoti, kaip jo veikimą pakeisti, pakreipti mums reikiama linkme.
Kita labai plati struktūrinės biologijos taikymo sritis – vaistų kūrimas. Kalbant labai apskritai, jei turime kokią nors mus puolančią bakteriją ar virusą, galime sukurti cheminį junginį, kuris slopintų kažkurio „užpuolikui“ būtino baltymo veikimą ir veiktų kaip vaistas. Baltymų su prijungtomis vaistų ar vaistų fragmentų molekulėmis struktūros leidžia geriau suprasti, kaip tie vaistai su baltymais – taikiniais sąveikauja ir kaip tą sąveiką galima dar pagerinti.
Geras pavyzdys – visai neseniai pasibaigusi SARS-CoV2 viruso pandemija. Jos metu įvairių šalių mokslininkai per rekordiškai trumpą laiką, naudodami struktūrinės biologijos metodus, nustatė visų viruso baltymų struktūras. Taigi ir potencialius vaistų taikinius. Kartu šios žinios pravertė ir kuriant vakcinas.
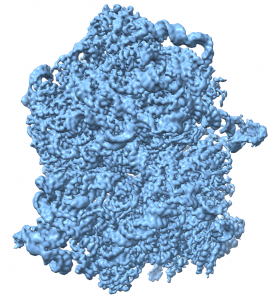
Cryo-EM map of a viral capsid
– Prieš pokalbį užsiminėte, kad turite pasidalinti ir savo naujausiu atradimu, sėkmės istorija. Papasakokite daugiau: ką pavyko atrasti?
– Kaip jau tikriausiai žinote, CRISPR-Cas9 ir kitos genų redagavimo žirklės yra mūsų „arkliukas“. Jos išgarsino Lietuvos mokslininkus pasaulyje, bet kartu ir puikiai padėjo populiarinti mokslą Lietuvos visuomenėje. O mes naudodami krio-EM sugebėjome nustatyti struktūrą baltymo, kuris yra CRISPR-Cas genomo redagavimo žirklių evoliucinis pirmtakas.
Šis baltymas, vadinamas TnpB, yra du ar tris kartus mažesnis ir kai kuriais atžvilgiais paprastesnis nei įprastos CRISPR-Cas žirklės. Pavasarį apie šią struktūrą paskelbėme straipsnį žurnale „Nature“. Mums tai labai svarbus straipsnis, tikra sėkmės istorija. Be to, tai – pirmoji publikuota struktūra, gauta mūsų krio-EM mikroskopu. Taip mes parodėme ir jo galimybes.
– Kodėl pradėjote ieškoti CRISPR-Cas pirmtako?
– Vadinamosios genomų žirklės, tai yra CRISPR-Cas9, CRISPR-Cas12 ir panašūs baltymai, yra randami bakterijose, jie padeda bakterijoms gintis nuo jas puolančių virusų, vadinamų bakteriofagais. Kilo klausimas, kaip atsirado tokie sudėtingi fermentai. Paaiškėjo, kad CRISPR žirklių pirmtakai atsirado transpozonais vadinamuose judriuosiuose genomo elementuose.
Kolegų atlikti tyrimai parodė, kad judriajame genomo elemente koduojamas baltymas, pavadintas TnpB, veikia labai panašiai kaip ir įprastos CRISPR genomo žirklės, bet yra gerokai mažesnis. Todėl mums rūpėjo sužinoti, kokia yra šių mažųjų „pirmykščių“ genomo žirklių sandara ir kaip jos veikia.
Tam, naudodami krio-EM, nustatėme TnpB baltymo struktūras. Pamatėme, kad nors TnpB baltymas yra daug mažesnis ir paprastesnis negu iš jo kilusios CRISPR žirklės, tai kažkiek kompensuoja su juo susijungusi didelė sudėtingos sandaros nukleorūgšties molekulė. Todėl TnpB atlieka tą pačią funkciją – tai yra atpažįsta tam tikrą DNR gabalą, jį suriša ir perkerpa.

Dr. Giedrius Sasnauskas
– Kaip manote, kaip žmonėms galėtų padėti jūsų ištirtas TnpB? CRISPR-Cas9, praėjus vos 12 metų nuo atradimo, jau yra taikomas gydant pjautuvinę anemiją, tyrimai tęsiasi ir toliau su kitomis genetinėmis ligomis. Taigi klinikinis pritaikymas atsirado labai greitai?
– Negaliu tiksliai pasakyti, kokie taikymo būdai bus atrasti vėliau. Genomų žirklių jau žinome kelis tūkstančius, populiariausios yra CRISPR-Cas9. Tačiau turime daug kitokių… Kad mokslininkai TnpB pradėtų taikyti, reikia jį nuodugniau charakterizuoti.
Pavyzdžiui, labai gerai, kad jis mažas, tai palengvina jo ar jo patobulintų variantų transportavimą į ląsteles. Tačiau kyla klausimas: ar jis toks pat tikslus kaip ir kiti įrankiai – ar pataiko ten, kur reikia? Jei pataiko kažkur ne ten, kur reikia, atsiranda ir naujos ligos rizika. Taigi iki taikymo praktikoje dar daug ką reikia išsiaiškinti.
– Man labai įdomu sužinoti jūsų nuomonę apie bendravimą su visuomene. Sakykime, šiandien aptartos sąvokos ir terminai, jeigu dauguma žmonių juos tiesiog kažkur perskaitytų, galbūt jiems nereikštų nieko. Bet kai papasakojate jūs, parodote, kiek daug visko vyksta, kaip mokslininkai nuolat plečia mūsų suvokimo ribas ir, kaip pats sakote, mato tai, ko nematė niekas iki šiol, tai tarsi įprasmina jas. Ar jūs pats bendraujate su visuomene? Ar manote, kad tai gali padėti mokslininkams apskritai?
– Manau, CRISPR-Cas9 išpopuliarinimas ir Lietuvos visuomenės susidomėjimas tikrai atnešė visokeriopą naudą. Mano indėlis į visuomenės švietimą iki šiol apsiribojo ekskursijomis, daugiausia skirtomis gamtos mokslais besidomintiems moksleiviams. Juos supažindinu su struktūrine biologija, pastaruoju metu – su krio-EM.
Platesnės visuomenės švietimu kol kas neužsiėmiau, buvau nuošalyje. Gal ir be reikalo, nes savo darbais pasidalinti su visuomene yra svarbu. Tačiau tai nėra lengva, gebėjimą komunikuoti reikia ugdyti. Juk techninės detalės ir mokslinio darbo rutina tikrai mažai kam įdomios. Reikia galvoti, kaip papasakoti tinkamai, kokius palyginimus sugalvoti ir pateikti, kad būtų visiems suprantamiau. Žodžiu, galbūt šis interviu bus puiki pradžia.
Kalbėjosi Goda Raibytė-Aleksa













Komentarų nėra. Būk pirmas!